【生醫評析】持續探求以AI/ML為基礎SaMD產品的最適化法規環境~FDA更新「軟體預先認證計畫」運作模式文件(v1.0)

在簡短介紹過美國政府所提出的國家AI發展政策及FDA 為確保利用「人工智慧」(Artificial intelligence,簡稱AI)或「機器學習」(Machine Learning,簡稱ML)技術所開發出的軟體醫療器材產品的安全性與功效所發布的「針對做為醫療器材使用之以AI/ML為基礎軟體(即SaMD)之法規管理架構修正草案-討論文件(Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD)-Discussion Paper)」後,這回筆者打算要來與讀者們分享一下,FDA 為探求並建構出對SaMD產品最適合的法規監管模式所提出的更新版「軟體預先認證計畫」(Software Pre-Certification Program)的運作模式文件(此為第3版),以更深入的瞭解該國主管機關,對於此種在技術與行政管理上較具複雜性與不確定風險的商品,現階段究係抱持著何種規範思維與態度?
有關FDA最初為何會提出「軟體預先認證計畫」的理由,主要是因認知到,其過往對「以硬體為基礎」(Hardware-Based)的醫療器材產品上市所採的傳統法規監管方法,對此類產品創新週期或變更頻率更快速、採循環式設計概念所開發的軟體醫療器材(包含以AI/ML為基礎的SaMD),已無法完全適用,如繼續強行套用傳統的法規監管模式,很可能將會阻礙未來病患近用此類以新創技術軟體為基礎的醫療產品的機會,故FDA方面決定,應有必要另行建構一套更加靈活且切合產品發展實務需求的法規監管模式。有鑑於此,於2017年7月時,FDA在以要求此類產品開發商能展現出一種穩健的「品質文化與組織機構卓越性」(Culture of Quality and Organizational Excellence,簡稱CQOE)並主動承諾將對其上市後產品的功能表現進行持續監控此等自律性規範理念為基礎的前提下,首次發布了「軟體預先認證計畫」(此計畫為試行性計畫)及相關配套文件,要嘗試藉由執行此項計畫所逐步累積的實務經驗及回饋意見,利用定期檢視與不斷更新計畫版本的方式,以一步步的形塑出一套能真正有效的因應此類產品上市後的相關問題並確保美國消費者能持續使用安全且有效產品的新法規監管模式。而根據目前筆者手邊最新版運作模式文件內容顯示,FDA於2018年4月26日,就「軟體預先認證計畫」的落實,即發布了第1版運作模式文件(即ver. 0.1),並於同年6月19日,迅速修正提出了第2版文件(即ver. 0.2),而後至2019年1月7日,在參酌回饋意見更細緻化相關管理措施前提下,再次修正並提出了第3版運作模式文件(ver. 1.0,此即本文重點)。
承上述,就本次所更新的第3版運作模式文件內容(不含附錄大致分8個部分),主要是FDA根據其自2018年中旬至同年10月31日此段期間所收集的回饋意見內容,針對幾個部分例如:新增對整體產品生命週期方法的詳細描述、調修對預先認證等級的說明、修正SaMD「產品-特定」分級要素、說明建構「真實世界功能表現分析計畫」(Real-World Performance Analytics Plan)相關流程等,由於整份文件涵蓋面向相當廣泛(且非首次提出),故以下,筆者將僅就更新版文件中較重要的修正部分,稍加整理與說明(對此計畫完整內容有興趣的讀者可透過參考資料網址自行至FDA官網下載閱讀):
(一)「軟體預先認證計畫」的整體概念說明部分
有關FDA最初為何會提出「軟體預先認證計畫」的理由,主要是因認知到,其過往對「以硬體為基礎」(Hardware-Based)的醫療器材產品上市所採的傳統法規監管方法,對此類產品創新週期或變更頻率更快速、採循環式設計概念所開發的軟體醫療器材(包含以AI/ML為基礎的SaMD),已無法完全適用,如繼續強行套用傳統的法規監管模式,很可能將會阻礙未來病患近用此類以新創技術軟體為基礎的醫療產品的機會,故FDA方面決定,應有必要另行建構一套更加靈活且切合產品發展實務需求的法規監管模式。有鑑於此,於2017年7月時,FDA在以要求此類產品開發商能展現出一種穩健的「品質文化與組織機構卓越性」(Culture of Quality and Organizational Excellence,簡稱CQOE)並主動承諾將對其上市後產品的功能表現進行持續監控此等自律性規範理念為基礎的前提下,首次發布了「軟體預先認證計畫」(此計畫為試行性計畫)及相關配套文件,要嘗試藉由執行此項計畫所逐步累積的實務經驗及回饋意見,利用定期檢視與不斷更新計畫版本的方式,以一步步的形塑出一套能真正有效的因應此類產品上市後的相關問題並確保美國消費者能持續使用安全且有效產品的新法規監管模式。而根據目前筆者手邊最新版運作模式文件內容顯示,FDA於2018年4月26日,就「軟體預先認證計畫」的落實,即發布了第1版運作模式文件(即ver. 0.1),並於同年6月19日,迅速修正提出了第2版文件(即ver. 0.2),而後至2019年1月7日,在參酌回饋意見更細緻化相關管理措施前提下,再次修正並提出了第3版運作模式文件(ver. 1.0,此即本文重點)。
承上述,就本次所更新的第3版運作模式文件內容(不含附錄大致分8個部分),主要是FDA根據其自2018年中旬至同年10月31日此段期間所收集的回饋意見內容,針對幾個部分例如:新增對整體產品生命週期方法的詳細描述、調修對預先認證等級的說明、修正SaMD「產品-特定」分級要素、說明建構「真實世界功能表現分析計畫」(Real-World Performance Analytics Plan)相關流程等,由於整份文件涵蓋面向相當廣泛(且非首次提出),故以下,筆者將僅就更新版文件中較重要的修正部分,稍加整理與說明(對此計畫完整內容有興趣的讀者可透過參考資料網址自行至FDA官網下載閱讀):
(一)「軟體預先認證計畫」的整體概念說明部分
1. 此計畫的基本概念,是基於對可展現出「品質文化與組織機構卓越性」的軟體製造商所開發的產品進行的預先性認證。
2. 首先,FDA將利用下列5項CQOE原則(又稱為「卓越性原則」)來評估某組織機構(即軟體產品開發商)本身的卓越性,包括:
2. 首先,FDA將利用下列5項CQOE原則(又稱為「卓越性原則」)來評估某組織機構(即軟體產品開發商)本身的卓越性,包括:
(1) 產品品質:在SaMD產品的開發、測試及維護等方面,表現出卓越的品質水準。
(2) 臨床責任:在「負責任的執行臨床評估」及「確保以病患為中心的議題可被適當處理(包括標示及人為因素)」等方面表現卓越。
(3) 網絡安全責任:在網路安全保護方面表現卓越,並藉由積極與利益相關者及同行間的互動溝通,來主動地解決網路安全相關問題。
(4) 病患安全:在提供安全的病患體驗及強調病患安全係為所有決策過程中的關鍵因素等方面表現卓越。
(5) 主動性文化:在主動監控、評估使用者需求及持續學習等方面表現卓越。
(2) 臨床責任:在「負責任的執行臨床評估」及「確保以病患為中心的議題可被適當處理(包括標示及人為因素)」等方面表現卓越。
(3) 網絡安全責任:在網路安全保護方面表現卓越,並藉由積極與利益相關者及同行間的互動溝通,來主動地解決網路安全相關問題。
(4) 病患安全:在提供安全的病患體驗及強調病患安全係為所有決策過程中的關鍵因素等方面表現卓越。
(5) 主動性文化:在主動監控、評估使用者需求及持續學習等方面表現卓越。
3. 其次,FDA將妥善運用從預先認證程序中所收集的數據資料,探求並採取一種「以風險為基礎」(Risk-Based)的、經簡化後的法規監管方法,來對SaMD產品進行審查,以藉此:
(1)取代傳統上對上市前提交相關文件的要求、或
(2)允許較高風險產品進行簡化上市前審查,來最大程度的提升審查效率與廠商參與度。
(2)允許較高風險產品進行簡化上市前審查,來最大程度的提升審查效率與廠商參與度。
4. 有關對於SaMD產品後續上市前審查途徑的判定,FDA說明將利用最小負擔原則,透過妥善利用產品於真實世界的功能表現資料,來衡平其於管理此類產品時,對SaMD產品上市前/後相關資訊的需求。
5. 而與FDA目前對以硬體為基礎的醫療器材產品所採的法規監管模式相類似的地方是…並非所有的醫療器材都需要進行上市前審查(例如:510(k)豁免醫療器材產品);因此,此項計畫預設,對由經預先認證的廠商所開發、提供及維護的「低風險」SaMD產品,原則上可豁免進行上市前審查,而對「高風險」SaMD產品,則將執行更有效率的上市前審查。
6. 質言之,按更新版運作模式文件內容,此項計畫乃是以要建構出一個具包容性、實用性及可評估任何組織機構(不論大企業或小型新創廠商)是否具有可展現其自身CQOE能力、最小化未來對此類軟體產品的行政管理負擔,並同時建立公眾信心(理由是因為經認證具CQOE能力的組織機構可以開發出具高品質的SaMD產品)的具彈性最適化管理模式為最終目標。
5. 而與FDA目前對以硬體為基礎的醫療器材產品所採的法規監管模式相類似的地方是…並非所有的醫療器材都需要進行上市前審查(例如:510(k)豁免醫療器材產品);因此,此項計畫預設,對由經預先認證的廠商所開發、提供及維護的「低風險」SaMD產品,原則上可豁免進行上市前審查,而對「高風險」SaMD產品,則將執行更有效率的上市前審查。
6. 質言之,按更新版運作模式文件內容,此項計畫乃是以要建構出一個具包容性、實用性及可評估任何組織機構(不論大企業或小型新創廠商)是否具有可展現其自身CQOE能力、最小化未來對此類軟體產品的行政管理負擔,並同時建立公眾信心(理由是因為經認證具CQOE能力的組織機構可以開發出具高品質的SaMD產品)的具彈性最適化管理模式為最終目標。
(二)計畫適用範圍的補充說明部分
1. 此文件指明,就正在開發或正規劃開發可能會受FDA法規監管的軟體產品的開發廠商(不論其規模大小),均可被納入此項預先認證計畫範圍,從表面上看,最終可以受益的產品類型,在概念上雖包含可符合FD&C Act, Section 201(h)規定中所描述的醫療器材定義的所有軟體產品(包含SaMD產品)、嵌入醫療器材中的軟體(Software in a Medical Device,簡稱SiMD)以及其他可被視為硬體醫療器材附加元件的軟體,不過,若再詳細檢視FDA目前所公布的更新版文件內容,可發現其乃將計畫執行重點,擺放在建立各種SaMD技術(或包含使用AI及ML演算法等軟體功能)的管理流程上,也就是說,就現階段計畫實際的適用範圍,實際上是僅限SaMD產品(關於「軟體即是醫療器材」【Software as a Medical Device,即SaMD】的定義為:是指就用於1種或多種醫療目的的軟體,可在不做為硬體醫療器材一部的情況下,單獨的實現那些醫療目的),以讓FDA可在預先認證計畫執行過程中,優先獲得SaMD產品的相關管理經驗。
2. 值得留意的是,若廠商所開發軟體產品功能是意圖供下列目的使用時,其非屬醫療器材,將不受FDA的法規監管,也不會落入此項計畫的範圍:
2. 值得留意的是,若廠商所開發軟體產品功能是意圖供下列目的使用時,其非屬醫療器材,將不受FDA的法規監管,也不會落入此項計畫的範圍:
(1) 為某種健康照護設施提供行政管理支援;
(2) 為維持或鼓勵健康的生活方式;
(3) 做為某些實際類型的電子病歷;
(4) 為傳輸、存儲、轉換格式或顯示數據資料而不解釋或分析臨床實驗室檢測或其他醫療器材的數據資料、結果及發現;或
(5) 為供某些有限型臨床決策支援等。
(2) 為維持或鼓勵健康的生活方式;
(3) 做為某些實際類型的電子病歷;
(4) 為傳輸、存儲、轉換格式或顯示數據資料而不解釋或分析臨床實驗室檢測或其他醫療器材的數據資料、結果及發現;或
(5) 為供某些有限型臨床決策支援等。
(三)對FDA未來將套用「產品整體生命週期」方法所為補充說明部分
1. 依更新版文件內容,指出可藉由將「產品整體生命週期」(即Total Product Lifecycle,簡稱TPLC)方法,套用至對軟體產品的法規監管流程的方式,來實現此計畫的目標及願景,其重點如後:
(1)於組織機構的「卓越性評估」(Excellence Appraisal)階段,將針對該開發商本身對「提供安全的病患體驗」所做的承諾及能力及其「考量病患安全」的優先順序(此即將病患安全議題做為該組織機構所有相關的決策-作成流程中的一項關鍵因素)等部分進行評估;
(2)要求開發商必須要對所開發產品的安全性進行持續性的監測與評估,以便在出現任何病患安全問題時,能儘早發現並迅速地進行補救。
(2)要求開發商必須要對所開發產品的安全性進行持續性的監測與評估,以便在出現任何病患安全問題時,能儘早發現並迅速地進行補救。
2. 為實現計畫目標,FDA將整部計畫拆分為如下4大Components(此為原始文件中的用詞,而筆者認為,不論中文轉譯為元件或組件,似均無法貼合原意,故直接援用Component一詞或較無疑慮),其依序為(如附圖一):
(1) Components-1:藉由卓越性評估方法來展現該組織機構的CQOE。
(2) Components-2:藉由審查判定機制來決定該項SaMD產品所需進行的審查方式。
(3) Components-3:執行簡化審查。
(4) Components-4:藉由產品上市後的真實世界的功能表現資料,來驗證該SaMD產品的持續的安全性、效用、功能表現和該組織機構對品質文化所為承諾。
(2) Components-2:藉由審查判定機制來決定該項SaMD產品所需進行的審查方式。
(3) Components-3:執行簡化審查。
(4) Components-4:藉由產品上市後的真實世界的功能表現資料,來驗證該SaMD產品的持續的安全性、效用、功能表現和該組織機構對品質文化所為承諾。
3. 更新版文件強調,就上開所列各項Component間,原則上彼此間存在著互相依存的關係。
附圖一、軟體預先認證計畫的4大項Components
.jpg)
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.12.
4. 另外,有關此種針對軟體產品將來的法規監管所設計的TPLC方法的實際概念,讀者們可參考本文附圖二說明,比較容易建立印象。而據更新版文件內容,其說明未來如導入TPLC方法進行管理的好處是,除可對此類軟體產品(包含具AI/ML功能軟體)從上市前的設計開發到上市後的功能表現進行持續的評估與監控外,更同時可持續展現該開發廠商的卓越性。
附圖二、軟體預先認證計畫的TPLC方法概念圖示
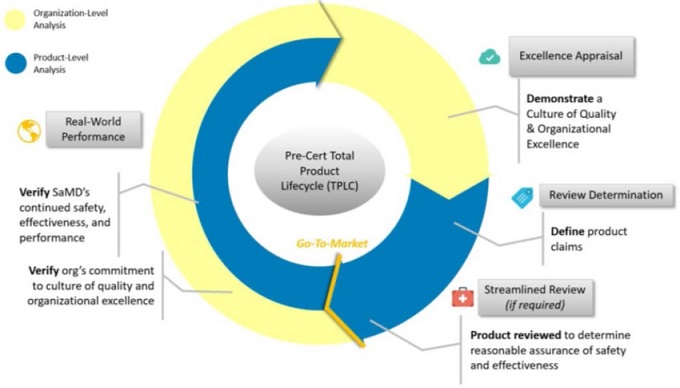
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.14.
附圖一、軟體預先認證計畫的4大項Components
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.12.
4. 另外,有關此種針對軟體產品將來的法規監管所設計的TPLC方法的實際概念,讀者們可參考本文附圖二說明,比較容易建立印象。而據更新版文件內容,其說明未來如導入TPLC方法進行管理的好處是,除可對此類軟體產品(包含具AI/ML功能軟體)從上市前的設計開發到上市後的功能表現進行持續的評估與監控外,更同時可持續展現該開發廠商的卓越性。
附圖二、軟體預先認證計畫的TPLC方法概念圖示
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.14.
(四)對SaMD產品開發廠商卓越性評估所提出的預先認證等級機制部分
1. 按更新版文件內容,依預先認證的目的及開發廠商本身的成熟度,FDA未來將建置2種不同的預先認證級別,略說明如下:
(1) 第1等級的預先認證:此等級的認證,目的是在允許能客觀展現其產品開發的卓越性,但在開發、交付及維護產品等方面「僅具有限記錄」的開發廠商,可在無需經審查的情況下,開發並銷售其某些具較低風險的軟體產品(但將要求該廠商其他類型的軟體產品進行簡化審查)。根據FDA方面的說法,此等級的認證,可能會對在交付軟體產品方面「僅具有限經驗」或「無任何經驗」,但其自身所採行的組織要素及策略,能確實展現出其具備(或能夠獲得)交付與維護安全/有效的低風險高品質的SaMD產品能力的開發廠商有利。
(2) 第2等級的預先認證:此等級的認證,目的是在允許能客觀展現其產品開發的卓越性,並在開發、交付及維護產品方面「具有良好記錄的」的開發廠商,可開發並銷售某些低風險及中度風險的軟體產品(且該廠商其他類型軟體產品可無須進行簡化審查)。根據文件說明,此等級的認證,可能對在交付軟體產品方面「具豐富經驗」的開發廠商有利。
(2) 第2等級的預先認證:此等級的認證,目的是在允許能客觀展現其產品開發的卓越性,並在開發、交付及維護產品方面「具有良好記錄的」的開發廠商,可開發並銷售某些低風險及中度風險的軟體產品(且該廠商其他類型軟體產品可無須進行簡化審查)。根據文件說明,此等級的認證,可能對在交付軟體產品方面「具豐富經驗」的開發廠商有利。
(五)對SaMD的「產品-特定」風險分級要素的細緻化描述部分
1. 為確定SaMD本身的風險等級,並以此為基礎續行判定該產品將來所需執行的上市前審查方式,FDA提出如下幾項關鍵要素,供開發廠商在製作SaMD產品聲明文件上參考:
(1) 聲明SaMD產品所提供資訊對醫療決定的重要性:即鑑別SaMD的預期醫療目的,也就是說,開發廠商應使用如下附表一中所列詞語,明確聲明其SaMD產品究如何可符合FD&C Act, Section 201(h)規定中所明定的醫療器材定義(如附表二所示),而就不符合醫療器材定義或可能符合醫療器材定義但FDA已表示將不打算強制其遵循得適用FD&C Act要求的SaMD產品,其開發廠商無需提供「SaMD產品所提供資訊對醫療決定的重要性」及其他相關資訊。
附表一、根據醫療狀況及該產品所提供資訊對醫療決策重要性的IMDRF-SaMD產品類型(即第I~IV類)判斷表
附表一、根據醫療狀況及該產品所提供資訊對醫療決策重要性的IMDRF-SaMD產品類型(即第I~IV類)判斷表
| 醫療狀況或條件 | 由SaMD所提供的資訊對醫療決策的重要性 | ||
| 治療或診斷 | 驅動臨床管理 | 為臨床管理提供資訊 | |
| 危急的 | IV | III | II |
| 嚴重的 | III | II | I |
| 不嚴重的 | II | I | I |
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.14.
附表二、SaMD產品所提供資訊對醫療決定的重要性聲明例示表
附表二、SaMD產品所提供資訊對醫療決定的重要性聲明例示表
| 資訊重要性的描述 |
|
為治療或診斷,例如:
- 為人體提供治療。
- 診斷/篩檢/檢測疾病或病徵。
驅動臨床管理,例如:
- 藉由提升支援藥品或醫療器材的安全及有效的使用,從而幫助治療。
- 為協助做出明確的診斷。
- 為檢傷分類或鑑別疾病或病徵的早期跡象。
為臨床管理提供資訊,例如:
- 告知各種選擇。
- 藉由匯整相關資訊以提供臨床資訊。
|
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.28.
(2) 聲明預期的醫療狀況或條件的狀態:此係指SaMD產品的預期用途,範圍包含預期的使用者、預期適用的疾病(或病徵)及預期適用的人口群等。開發廠商可以參考下述附表三來撰寫此部分聲明。就不符合醫療器材定義或可能符合醫療器材定義但FDA已表示不打算強制其遵循得適用FD&C Act要求的SaMD產品,開發廠商無需提供其「醫療狀況或條件的狀態」此部分資訊。
附表三、SaMD產品醫療狀況或條件的狀態聲明用詞範例表
(2) 聲明預期的醫療狀況或條件的狀態:此係指SaMD產品的預期用途,範圍包含預期的使用者、預期適用的疾病(或病徵)及預期適用的人口群等。開發廠商可以參考下述附表三來撰寫此部分聲明。就不符合醫療器材定義或可能符合醫療器材定義但FDA已表示不打算強制其遵循得適用FD&C Act要求的SaMD產品,開發廠商無需提供其「醫療狀況或條件的狀態」此部分資訊。
附表三、SaMD產品醫療狀況或條件的狀態聲明用詞範例表
| 危急 | 嚴重 | 非嚴重 | |
|---|---|---|---|
| 疾病或病徵的類型為: | - 健康的威脅生命狀態,包括無法治癒的狀態。 - 需要重大的治療介入。 - 有時時間急迫,取決於可能影響使用者反映於輸出資訊能力的疾病或病徵的病程發展狀況。 |
- 進展溫和,多半可治癒。 - 無需重大治療介入。 - 介入通常不被預期為避免死亡、長期殘疾或其他嚴重的健康惡化而具時間急迫性,以避免死亡,而因此讓使用者有能力可發現錯誤的建議。 |
- 緩慢的、伴隨可預測的疾病狀態的病程發(可能包含輕微的慢性疾病或狀態)。 - 或可能無法被治癒,但可有效的加以管理。 - 只需要輕微的治療介入。 - 介入措施通常是非侵入性的,可讓使用者有能力發現錯誤的建議。 |
| 預期的目標人群為: | 易受傷害的疾病或病徵人群(例如:兒童、高風險人群等)。 | 非易受傷害的疾病或病徵人群。 | 並不一定是病患的個體。 |
| 預期將會被那些人員使用: | 經專業培訓的使用者。 | 經專業訓練的使用者或一般使用者。 | 經專業訓練的使用者或一般使用者。 |
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.28.
(3) 聲明SaMD產品的核心功能:亦即應鑑別其SaMD產品本身所具的關鍵性特徵/功能,而此處所謂的關鍵性特徵/功能,是指在所預期的醫療情況下,對該項SaMD產品所提供資訊對醫療上相關決定-作成具關鍵性影響的那些特徵或功能。故就此部分提交的說明,宜包含:可維持功能表現、安全性側寫與(或)其他特徵的特定功能等資訊。
(4) 就廠商針對其醫療器材所為的描述中,原則上可包含對該項軟體醫療器材的一般性描述,例如:解釋軟體如何運作、重大的安全/技術與防護風險、與支援平台/元件及相容性有關的資訊、使用說明&限制、所使用的輸入資料以及消費者支援等。
(5) 至廠商針對其SaMD產品的功能表現所為的說明,則可包含對該項軟體醫療器材的功能表現特徵的一般性描述,例如:SaMD在預期的醫療情況或條件下的分析或其臨床功能表現、與該SaMD產品在隱私保護&安全性政策方面有關的資訊等。
(6) 綜合上開5項資訊,於同時納入:i.開發廠商的預先認證等級(第1等級&第2等級)與ii.該項SaMD產品的風險類別(等級I至等級IV)前提下,就某SaMD產品未來宜採何種上市前審查途徑(或模式),基本上可產生下列兩種判斷結果(可參看附表四),亦即:
(3) 聲明SaMD產品的核心功能:亦即應鑑別其SaMD產品本身所具的關鍵性特徵/功能,而此處所謂的關鍵性特徵/功能,是指在所預期的醫療情況下,對該項SaMD產品所提供資訊對醫療上相關決定-作成具關鍵性影響的那些特徵或功能。故就此部分提交的說明,宜包含:可維持功能表現、安全性側寫與(或)其他特徵的特定功能等資訊。
(4) 就廠商針對其醫療器材所為的描述中,原則上可包含對該項軟體醫療器材的一般性描述,例如:解釋軟體如何運作、重大的安全/技術與防護風險、與支援平台/元件及相容性有關的資訊、使用說明&限制、所使用的輸入資料以及消費者支援等。
(5) 至廠商針對其SaMD產品的功能表現所為的說明,則可包含對該項軟體醫療器材的功能表現特徵的一般性描述,例如:SaMD在預期的醫療情況或條件下的分析或其臨床功能表現、與該SaMD產品在隱私保護&安全性政策方面有關的資訊等。
(6) 綜合上開5項資訊,於同時納入:i.開發廠商的預先認證等級(第1等級&第2等級)與ii.該項SaMD產品的風險類別(等級I至等級IV)前提下,就某SaMD產品未來宜採何種上市前審查途徑(或模式),基本上可產生下列兩種判斷結果(可參看附表四),亦即:
- 無須進行上市前審查(No Review)或
- 須執行簡化上市前審查(SR)
- 須執行簡化上市前審查(SR)
附表四、於未來預先認證計畫中FDA就取得第1與第2預先認證等級組織機構開發的SaMD產品所提議可採的審查程度判斷表
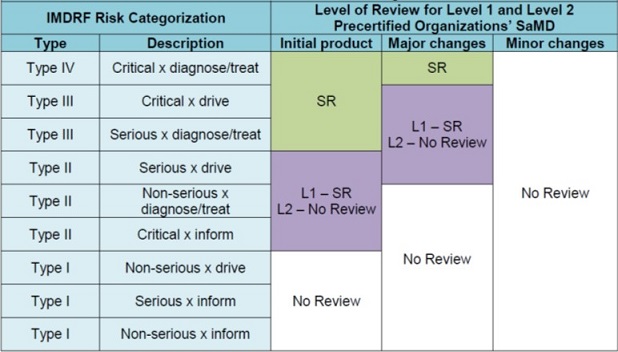
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.29.
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.29.
(六)對軟體預先認證計畫中的簡化上市審查流程所為補充說明部分
1. 有關此項軟體預先認證計畫文件中所提出的簡化上市前審查,將會提供給那些於「真實世界的功能表現分析」方面可展現卓越能力與持續承諾的經預先認證的開發廠商利用;而事實上,前述簡化上市審查程序的目標,主要是欲鑑別出SaMD產品於上市前審查所需的關鍵要素,並為由經預先認證的廠商所開發此類產品的安全及有效性提供合理保證。
2. 原則上,針對上述簡化審查程序FDA預計應可從如下幾個面向發揮作用:
2. 原則上,針對上述簡化審查程序FDA預計應可從如下幾個面向發揮作用:
(1) 於充分瞭解產品方面:主管機關將妥善利用開發廠商於審查途徑判定階段所預先提交的資訊(如有),並將會與計畫參與者進行互動合作,以充分瞭解該項軟體產品功能等相關細節。
(2) 於上市前審查方面:主管機關將會對支援性資料執行互動式審查(包括評估該軟體的分析功能表現、臨床功能表現及適當的安全措施)。
(3) 於上市授權方面:主管機關將會作成上市前決定、文件化決定摘要、保留作成決定所依據的資料與記錄,並將其決定傳達給該開發廠商。
(2) 於上市前審查方面:主管機關將會對支援性資料執行互動式審查(包括評估該軟體的分析功能表現、臨床功能表現及適當的安全措施)。
(3) 於上市授權方面:主管機關將會作成上市前決定、文件化決定摘要、保留作成決定所依據的資料與記錄,並將其決定傳達給該開發廠商。
3. 按更新版文件內容,就傳統規範架構下廠商於SaMD產品上市前審查時所提交文件中所包含的某些要素(Elements),FDA認為可在廠商卓越性評估階段中,從「組織機構」的層級來進行評估,而至上市前審查途徑判定階段,則可從「產品」的層次上來進行評估。
4. 就上開於更新版文件中所提議可做為確保SaMD產品安全性與有效性所必需加以評估的「產品-特定」(Product-Specific)關鍵要素,略說明於下:
4. 就上開於更新版文件中所提議可做為確保SaMD產品安全性與有效性所必需加以評估的「產品-特定」(Product-Specific)關鍵要素,略說明於下:
(1) 臨床演算法(包括作用機制、設計、開發):此項要素應充分提供任何與所使用的臨床演算法有關的資訊,包括作用機制。
(2) 臨床功能表現:此要素應包含臨床資料分析及解釋,以證明該醫療器材確實具有適當的臨床功能表現。
(3) 網路安全:在進行廠商本身的卓越性評估階段,FDA將從評估安全、管制更新、對其產品執行上市後安全評估及主動更新產品的能力等方面來評估廠商能力。而於簡化審查階段,FDA將針對「產品-特定」的網路安全要素進行評估,包括經鑑別出的威脅及緩解措施將如何影響醫療器材的安全性。
(4) 危害分析:鑑於「產品-特定」的風險、危害及後續的緩解措施,對瞭解醫療器材預期用途的安全及有效性至關重要,故FDA將其設定為簡化審查要素的一部。
(5) 使用說明/標籤:此要素應包括可協助使用者瞭解&如何使用醫療器材的說明或其他材料(電子化文件或其他)。
(6) 法規監管途徑特定項目:簡化審查資料將包含如510(k)所提交的實質等同(SE)比較資料以鑑別出該產品本身屬性、解釋所鑑別出的特定管制措施如何可滿足法規要求、或風險/效益分析等。
(7) 修正歷程:修正歷程是為該項醫療器材特定的,因其可協助審查人員瞭解於設計過程中任何可能影響該產品的安全及有效性的「產品-特定」變更。(經簡化審查的低風險軟體產品不包含此項要素,因為其可藉由卓越性評估中對軟體開發流程所為的評估來充分的加以證明(對低風險產品)。
(8) 軟體架構:此項要素係指可就功能單元部分與軟體模組提供詳細的說明資料,或可能包括狀態圖(State Diagrams)及流程圖。
(9) 驗證(功能表現訴求/醫療器材功能表現):於此項要素中,由於臨床演算法的驗證是最為重要的,故FDA提議應全面性的加以說明(包括供測試用的協定與展示其功能表現結果),而對於較低風險的SaMD產品,FDA可以接受產商提交驗證資訊的摘要資料)。
(2) 臨床功能表現:此要素應包含臨床資料分析及解釋,以證明該醫療器材確實具有適當的臨床功能表現。
(3) 網路安全:在進行廠商本身的卓越性評估階段,FDA將從評估安全、管制更新、對其產品執行上市後安全評估及主動更新產品的能力等方面來評估廠商能力。而於簡化審查階段,FDA將針對「產品-特定」的網路安全要素進行評估,包括經鑑別出的威脅及緩解措施將如何影響醫療器材的安全性。
(4) 危害分析:鑑於「產品-特定」的風險、危害及後續的緩解措施,對瞭解醫療器材預期用途的安全及有效性至關重要,故FDA將其設定為簡化審查要素的一部。
(5) 使用說明/標籤:此要素應包括可協助使用者瞭解&如何使用醫療器材的說明或其他材料(電子化文件或其他)。
(6) 法規監管途徑特定項目:簡化審查資料將包含如510(k)所提交的實質等同(SE)比較資料以鑑別出該產品本身屬性、解釋所鑑別出的特定管制措施如何可滿足法規要求、或風險/效益分析等。
(7) 修正歷程:修正歷程是為該項醫療器材特定的,因其可協助審查人員瞭解於設計過程中任何可能影響該產品的安全及有效性的「產品-特定」變更。(經簡化審查的低風險軟體產品不包含此項要素,因為其可藉由卓越性評估中對軟體開發流程所為的評估來充分的加以證明(對低風險產品)。
(8) 軟體架構:此項要素係指可就功能單元部分與軟體模組提供詳細的說明資料,或可能包括狀態圖(State Diagrams)及流程圖。
(9) 驗證(功能表現訴求/醫療器材功能表現):於此項要素中,由於臨床演算法的驗證是最為重要的,故FDA提議應全面性的加以說明(包括供測試用的協定與展示其功能表現結果),而對於較低風險的SaMD產品,FDA可以接受產商提交驗證資訊的摘要資料)。
5. 有關互動式簡化上市前審查程序的實際概念,讀者們可參考附圖三。
附圖三、互動式簡化上市前審查程序架構概念圖示
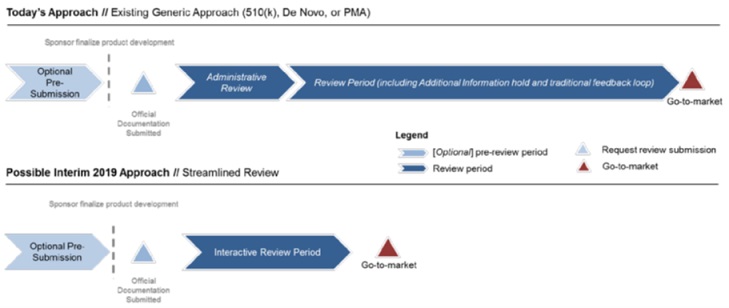
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.34.
附圖三、互動式簡化上市前審查程序架構概念圖示
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.34.
(七)針對SaMD產品的「真實世界的功能表現分析計畫」所為補充說明部分
1. 依更新版文件內容,有關軟體預先認證計畫中所描述的「真實世界的功能表現」(Real-World Performance)此部分,其主要目的是要妥善利用並分析那些既有資料,來驗證預先認證後開發廠商持續的卓越性並鑑別新出現的安全問題及網路安全風險,並為計畫的其他部分提供關鍵性回饋。
2. 就更新版文件中所稱「真實世界的功能表現的分析方法」(Real-World Performance Analytics,即RWPA),係定義為:「對由經預先認證的廠商所開發並已上市的SaMD產品的安全性、有效性及功能表現等存在於真實世界環境中的相關資料所執行的系統性計算分析」,且至少包括如下3類方法(可參考附圖四),即:
2. 就更新版文件中所稱「真實世界的功能表現的分析方法」(Real-World Performance Analytics,即RWPA),係定義為:「對由經預先認證的廠商所開發並已上市的SaMD產品的安全性、有效性及功能表現等存在於真實世界環境中的相關資料所執行的系統性計算分析」,且至少包括如下3類方法(可參考附圖四),即:
(1) 真實世界健康分析(RWHA):是指與SaMD產品的預期用途相關的真實世界的臨床產出及結果的分析。
(2) 使用者體驗分析方法(User Experience Analytics,簡稱UXA):係指與SaMD產品實際使用相關的使用者體驗輸出的分析方法。
(3) 產品功能表現分析方法(Product Performance Analytics,簡稱PPA):係為針對輸出與結果的分析,用以證明SaMD產品在真實世界中的準確性、可靠性及安全性。
(2) 使用者體驗分析方法(User Experience Analytics,簡稱UXA):係指與SaMD產品實際使用相關的使用者體驗輸出的分析方法。
(3) 產品功能表現分析方法(Product Performance Analytics,簡稱PPA):係為針對輸出與結果的分析,用以證明SaMD產品在真實世界中的準確性、可靠性及安全性。
附圖四、RWPA於產品上市後的監控領域示意圖
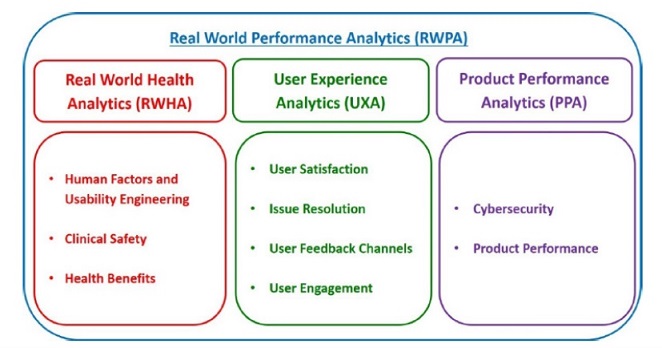
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.40.
參考資料:“Developing a Software Precertification Program:A working model-v1.0-January 2019”, p.40.
整體來說,FDA對此類具備「持續學習」與「不斷自我更新功能」的以AI/ML為基礎的SaMD產品將來的管理,其實看得出是確實下了不少工夫,除了積極的鼓勵公眾對其所公布每一版更新後的計畫內容提供回饋意見外,更是一件件的去實質檢視所收到的回饋意見並作成回應(對FDA所作成回應內容有興趣的讀者們可參看更新版文件的Appendix 9部分),並將其認為有價值的意見或機制,逐一納入未來將更新的文件當中(於本質上這也就是一種結合公眾參與機制的滾動式修正方法),以讓該國SaMD產品未來蓬勃發展所需的法規環境更臻完善。事實上,我們如再回過頭來檢視這套帶有濃厚試行性色彩的軟體預先認證計畫,理論上應不難發覺此計畫本身,實蘊含了幾種規範思維,簡單的來說,就是:第一、是以保障品質及維護病患安全等核心價值,貫穿整部計畫;而第二、則是將管理重點,化約為兩部分,第一部分是,將開發廠商本身品質文化水準高低與是否將病患安全納入內部「決策-作成」流程,運用「組織機構的卓越性」這項概念來加以包裹,要企圖將FDA過往對醫療器材軟體產品所採的傳統法規監管模式,向前延伸至開發廠商本身(也就是組織機構層級的自律性管理),以更深一層的為將來對此類SaMD產品的品質與安全性管理,扎下根基;而至於第二部分,當然就是聚焦在對SaMD產品本身的安全性、功效及上市後風險的管理上,只不過…在這第二部分的管理過程當中,FDA特別運用了其在第一部分對開發廠商本身卓越性評估後的結果(也就是是否能滿足卓越性要求而成為FDA所認可的卓越組織機構),以及在評估過程中由開發廠商所預先提交的與SaMD產品有關的文件資料,來作為後續其SaMD產品可免審查、可進行簡化上市審查、達到縮減審查時程、減輕行政負擔及獲取開發廠商在產品上市後主動監測其產品的真實世界功能表現資料等事項的有力基礎。
截至目前為止,根據FDA於官網所公布的資料顯示,FDA已選出了包括:Apple、Fitbit、Johnson & Johnson、Pear Therapeutics、Phosphorus、Roche、Samsung、Tidepool、Verily等多家知名廠商來參與此項試行性軟體預先認證計畫(原計有100家機構申請參與),且在主管機關尚未正式提出SaMD新法規監管架構前,業已核准了超過20項以AI/ML為基礎的醫療器材軟體產品,例如:可獨立診斷出糖尿病所造成的視網膜病變的IDX-DR、或藉分析腦部電腦斷層影像來檢測腦部可疑的血管阻塞情況的ContaCT等,故就美國境內以AI/ML為基礎的SaMD產品的未來發展,在其國家AI政策的推波助瀾及民間企業的共襄盛舉下,待FDA完善其法規環境後,勢將大幅激勵相關領域廠商投入創新與開發此類產品的意願,而這點或也是值得讓外界抱持高度期待的另一項重要原因之一。
截至目前為止,根據FDA於官網所公布的資料顯示,FDA已選出了包括:Apple、Fitbit、Johnson & Johnson、Pear Therapeutics、Phosphorus、Roche、Samsung、Tidepool、Verily等多家知名廠商來參與此項試行性軟體預先認證計畫(原計有100家機構申請參與),且在主管機關尚未正式提出SaMD新法規監管架構前,業已核准了超過20項以AI/ML為基礎的醫療器材軟體產品,例如:可獨立診斷出糖尿病所造成的視網膜病變的IDX-DR、或藉分析腦部電腦斷層影像來檢測腦部可疑的血管阻塞情況的ContaCT等,故就美國境內以AI/ML為基礎的SaMD產品的未來發展,在其國家AI政策的推波助瀾及民間企業的共襄盛舉下,待FDA完善其法規環境後,勢將大幅激勵相關領域廠商投入創新與開發此類產品的意願,而這點或也是值得讓外界抱持高度期待的另一項重要原因之一。
參考資料
1. About the full text of the “Developing a Software Precertification Program:A working model-v1.0-January 2019”,
available at this website:https://www.fda.gov/media/119722/download
2. For more information about the “Digital Health Software Precertification (Pre-Cert) Program”,
please refer to this website:https://www.fda.gov/medical-devices/digital-health/digital-health-software-precertification-pre-cert-program
3. For more information about the “Global Approach to Software as a Medical Device Software as a Medical Device”,
please refer to this website:https://www.fda.gov/medical-devices/software-medical-device-samd/global-approach-software-medical-device-software-medical-device
4. For more information about the “Who Is Currently Involved in the Pre-Cert Pilot Program?”,
please refer to this website:https://www.fda.gov/medical-devices/digital-health/digital-health-software-precertification-pre-cert-program
5. About the full text of the “FDA's Digital Health Innovation Action Plan”,
available at this website:https://www.fda.gov/media/106331/download
6. 有關「打造人工智慧的美麗新願景-美國AI政策之回顧整理-I」系列文章,
可至巨群網站參考:http://www.giant-group.com.tw/law-detail-899.html
7. 有關「量身打造以AI技術為基礎之醫療器材軟體上市法規環境~美國FDA公布新管理架構概念討論文件」文章,
可至巨群網站參考:http://www.giant-group.com.tw/law-detail-734.html

