【生醫評析】美國醫療軟體規範翻修-Part-IV:FDA新修正版「於醫療器材中使用現成軟體」指引文件重點概覽
關於上述新修正版指引文件內容,包含附錄部分,大致可分7個章節,除適用範圍與重要用詞外,FDA花了較多的篇幅,來描述對使(採)用OTS軟體時所使用的危害性分析方法,並同時簡短的提供幾個使(採)用OTS軟體的醫療器材的例示性說明案例給製造商(或開發人員)參考,且在最末段章節中,FDA更另就幾項常見廠商提問,提供了重要的回應與官方解釋,故以下,筆者將針對前述部分中的幾個較值得觀察的地方,為讀者們進行整理,現說明於下:
(一) 新修正指引文件的適用範圍為何?
-
按FDA說明,其之所以修訂這份文件的目的,主要是為了說明對於涉及使用OTS 軟體的醫療器材,於其上市申請的過程中,廠商通常應準備與提供的資料有哪些?而這些通常會被FDA要求提供的資料,在性質上,其實是針對在這份名為“Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices”指引文件中所提到的那些個文件資料的一種「補充」資料。
-
關於在這份指引文件中所提到的數項原則,有助於製造商針對所開發醫療器材中使(採)用的OTS軟體,建立相應合適的「設計控制」與「確效計畫」。
-
值得注意的是,在這整份文件中,FDA乃廣泛使用了「危害分析」這個詞彙,而依FDA說明,這主要是為了強化如後概念,亦即:「在臨床上,如僅單純的以軟體的故障率來估算風險,一般而言並不是合理的,故應該要以『危害的嚴重程度』而不是以『軟體的故障率』來規範所使(採)用OTS軟體的安全風險,方較合適」。
(二) 有哪些重要用詞/概念?
(1) 如果與醫療器材功能相關的軟體的操作,將「直接」影響到病患、操作者與(或)周圍人員,其發生故障或潛在的設計缺陷,可能導致病患、操作者與(或)周圍人員受到“non-Serious”的傷害,或者...
(2) 如果與醫療器材功能相關的軟體的操作,將「間接」影響到病患、操作者與(或)周圍人員(如透過照護提供者的行為),其發生故障或潛在的設計缺陷(如當不正確或延遲的資訊),可能對病患、操作者與(或)周圍人員造成non-Serious的傷害。
(1) 如果與醫療器材功能相關的軟體的操作,將「直接」影響病患、操作者與(或)周圍人員,其故障或潛在缺陷,可能導致病患、操作者與(或)周圍人員「死亡或嚴重傷害」,或...
(2) 如果與醫療器材功能相關的軟體的操作,將「間接」影響病患、操作者與(或)周圍人員(如透過照護提供者的行為),其發生故障或潛在的設計缺陷(如當不正確或延遲的資訊),可能導致病患、操作者與(或)周圍人員「死亡或嚴重傷害」。
(三) FDA對用於醫療器材的OTS軟體將會考量哪些基本因素?
Q1:它(即OTS軟體)是甚麼?
對於在醫療器材產品(或系統)中所使用的每一項OTS軟體,FDA將會進一步要求廠商就後述幾個項目詳細加以說明,例如:OTS軟體的名稱及其原始製造商、軟體的版本級別、發佈日期、修補程式編號與升級名稱(如合適)、為什麼這個OTS軟體適用於該項醫療器材等等。
Q2:所使(採)用的OTS軟體其對應的電腦系統規格為何?
這個問題是指,就廠商所使(採)用的OTS軟體,其是在甚麼樣的組態(Configuration)下來進行確效(Validation)?
Q3:廠商將如何確保最終使用者將會採取適當的行動?
例如OTS軟體及電腦系統的哪些方面可以(與/或必須)安裝/設定組態?或者在該項醫療器材中有設計了哪些措施來防止運行任何non-Specific OTS軟體(例如文字處理器、遊戲軟體的非預期安裝)?
Q4:所使(採)用的OTS軟體擬發揮的作用為何?
這個問題是指,用於醫療器材產品(或系統)中的OTS軟體,其究竟將提供何種功能?
Q5:廠商是如何知道它是有效的?
這項問題是指,廠商應根據其所使(採)用的OTS軟體的「關注程度」(Level of Concern),詳細說明如後項目,例如:描述OTS軟體的測試、驗證與確效,並確保其與醫療器材危害相關的OTS 軟體間的合適性、提供測試的結果等、是否有目前最新的OTS軟體問題(bug)清單及更新途徑等。
Q6:廠商將如何保持對OTS軟體的追蹤(或管控)?
(四) 什麼是OTS軟體的「危害分析」?
-
依FDA所做說明,在這類型醫療器材產品的整體產品生命週期中,宜採取一種全面性的風險管理方法,並應持續的進行「危害分析」與「緩減措施」,而在這樣的前提下,由於廠商所使(採)用的OTS 軟體一旦失效、故障或錯誤使用,可能會對病患、操作者或周圍人員造成危害,因此,這類型醫療器材產品的製造商,多半會被FDA期待要執行可作為「整體醫療器材(系統)危害分析」的一部的「OTS軟體危害分析」,並檢附相關資料。
-
按上述FDA的期望,於廠商所提繳的「整體醫療器材(系統)危害分析」文件中,原則上還應要包含「OTS軟體的危害分析」的資料,而簡單的來說,這些資料,其實也就是指廠商針對如下項目所加以收集/紀錄並進行文件化的資訊,包括:
2. 每項已被鑑別出的潛在危害的嚴重程度估算;
3. 可能導致已被鑑別出的潛在危害的所有潛在原因清單。
(五) 什麼是OTS軟體的「危害緩減措施」?
-
按FDA建議,就廠商所為的危害緩減的相關活動,可以朝尋求降低危害的嚴重程度、發生的可能性,或兩者的方向進行規劃設計與執行。而原則上,前述危害緩減措施,依效用高低,大致可分為如下3類:
2. 保護措施(被動性措施);
3. 警告使用者(標示)。
-
上述所列的3種緩解方法,其並非相互排斥的,也就是說,就這3種方法廠商可以同時的加以運用。對此,FDA指出,最理想的方式,是設計有效的管控措施,也就是廠商主動的消除具危害性操作、或對具危害性元件的需求;至於採取保護性措施,FDA認為是較為被動的方法(這是從使用者的角度出發來進行考量),因為它們並不需要使用者採取任何行動;而至於單純的警告標示,則會被FDA認為是比較不有效的方法(指與前2種方法相比較),因為這種方法必須取決於使用者是否主動地採取(或不採取)某些行動。
-
就廠商所提繳的有關OTS軟體的危害緩解情況的文件化紀錄資料,FDA建議宜包含下述各項資訊:
2. 為減輕每種危害所採取的相關步驟與措施;以及
3. 殘餘風險。
(六) 什麼是OTS軟體的「殘餘風險」?
-
依FDA說明,對於廠商已採取危害緩解措施後尚存有的風險,建議宜在其所提交的文件資料中詳細加以描述及說明,且就「與其所使(採)用OTS軟體有關」的風險,亦應與替代方案(例如客製化開發的軟體)的風險,一同加以考慮。
-
而所謂可接受的殘餘風險程度,主要是在以「殘餘風險的嚴重性」或「發生殘餘風險的可能性」為基礎的前提下,視該項醫療器材的「預期用途」與「軟體所執行的功能」來判定。
-
此處需留意的是,此類醫療器材申請者,需就具“Moderate LoC”或“Major LoC”的「殘餘風險」,詳細的加以描述說明,且若廠商所使(採)用的 OTS 軟體,採取危害緩減後措施的殘餘風險,尚仍將構成Major LoC時,則FDA將要求廠商,應於開發過程中執行特殊的文件化紀錄。
(一) FDA的OTS軟體判斷流程
關於FDA對廠商所使(採)用的OTS軟體所為的風險評估判斷,可綜整為如下流程圖:
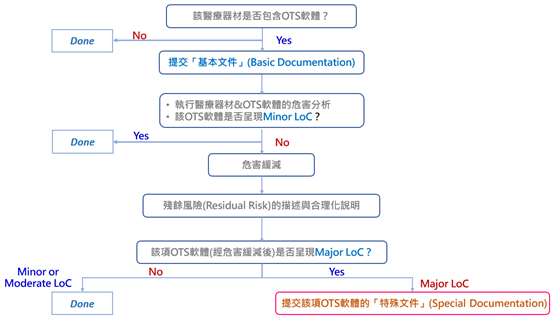
參考資料:“Off-The-Shelf Software Use in Medical Devices-Guidance for Industry and Food and Drug Administration Staff (2019)”, p.5。
(二) FDA建議製造商所提交的OTS軟體文件宜包含的內容
在以上述第(一)項流程圖為基礎下,就FDA建議於廠商提交的有關OTS軟體文件中所應該包含的資料,可整理出如下摘要表:
|
OTS軟體提交文件摘要表
|
||||||||||||||||||||||||||||
(補充說明:關於在這份文件後段所提到的幾項「例示性說明性案例」、及FDA對於廠商常見提問所做的官方回應等部分,雖礙於篇幅限制而未多作著墨,但筆者認為,這部分內容或多或少也具有相當程度的參考價值,故建議有餘裕的讀者們,不妨可一併瀏覽一下,相信應會有所收穫…。)
參考資料
1. For more information about the “FDA Guidance on Off-The-Shelf Software Use in Medical Devices”, please refer to this website:
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-use-medical-devices
2. About the full text of the “FDA Guidance on Off-The-Shelf Software Use in Medical Devices”, available at this website:
https://www.fda.gov/media/71794/download
3. For more information about the “FDA Guidance on Off-The-Shelf Software Use in Medical Devices”, please refer to this website:
https://www.regdesk.co/fda-guidance-on-off-the-shelf-software-use-in-medical-devices/
4. For more information about the “Deciding When to Submit a 510(k) for a Change to an Existing Device”, please refer to this website:
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-change-existing-device
5. For more information about the “Deciding When to Submit a 510(k) for a Software Change to an Existing Device”, please refer to this website:
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-software-change-existing-device

